Research
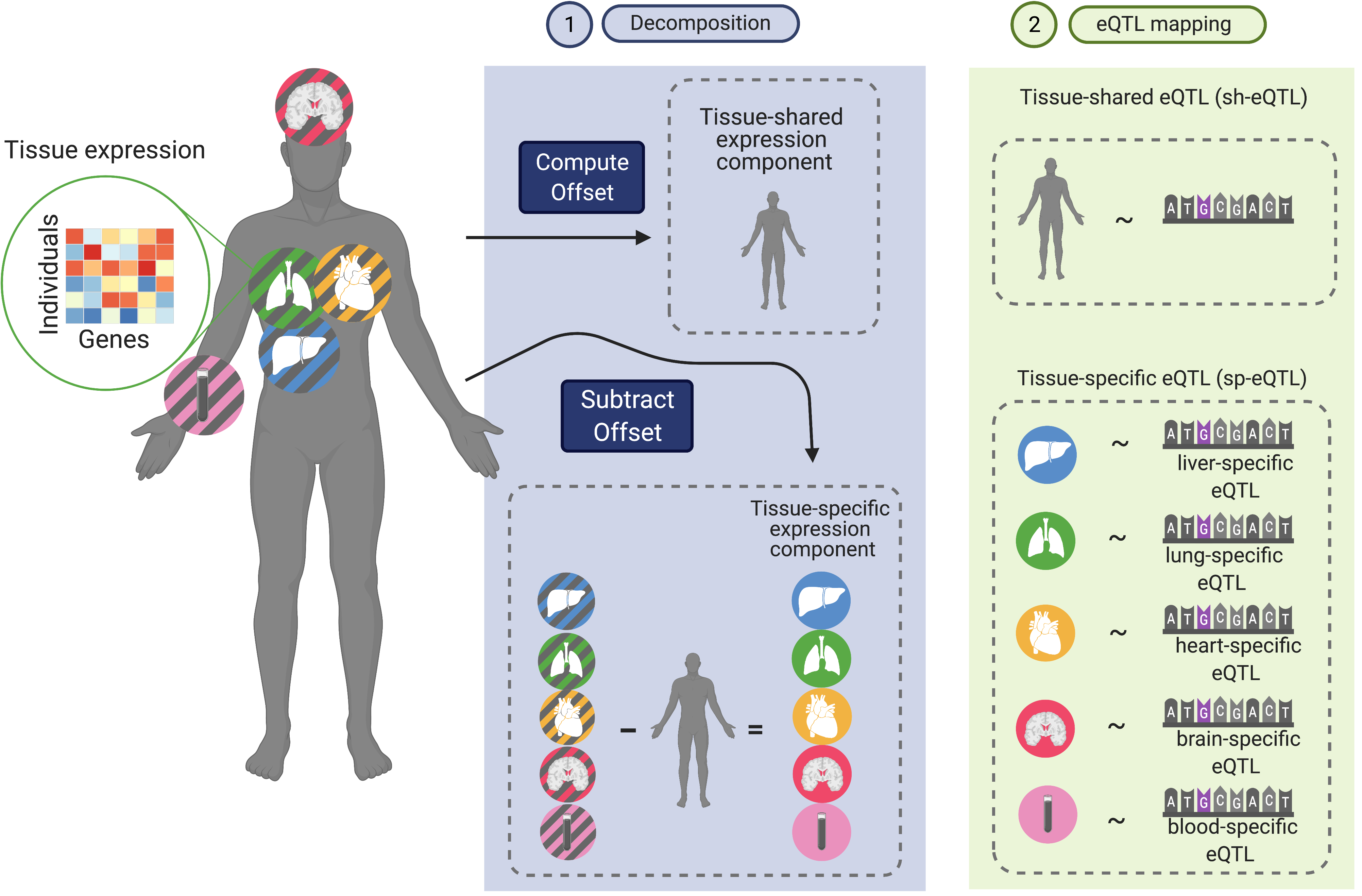
Mapping context-dependent regulatory effects
The vast majority of the loci identified from genetic association studies are located outside coding regions, complicating their mechanistic understanding. Our lab develops statistical methods and computational tools for contextualizing genetic associations by leveraging large-scale multi-context functional genomic data (e.g., Genotype-Tissue Expression Project, Human Cell Atlas) to find disease-relevant genes and contexts for disease-associated variants. Specifically, we have recently developed FastGxC, a computationally efficient and statistically powerful method for detecting context-specific QTL effects in multi-context genomic studies with shared noise. We have applied FastGxC to bulk multi-tissue and single-cell RNA-seq data sets to produce the most comprehensive tissue- and cell-type-specific eQTL maps to date. Together with collaborators at UCLA, we have also developed CONTENT, an extension of FastGxC to cross-context transcriptome-wide association studies to increase power to identify genes associated with complex traits and their tissue and cell types of action.
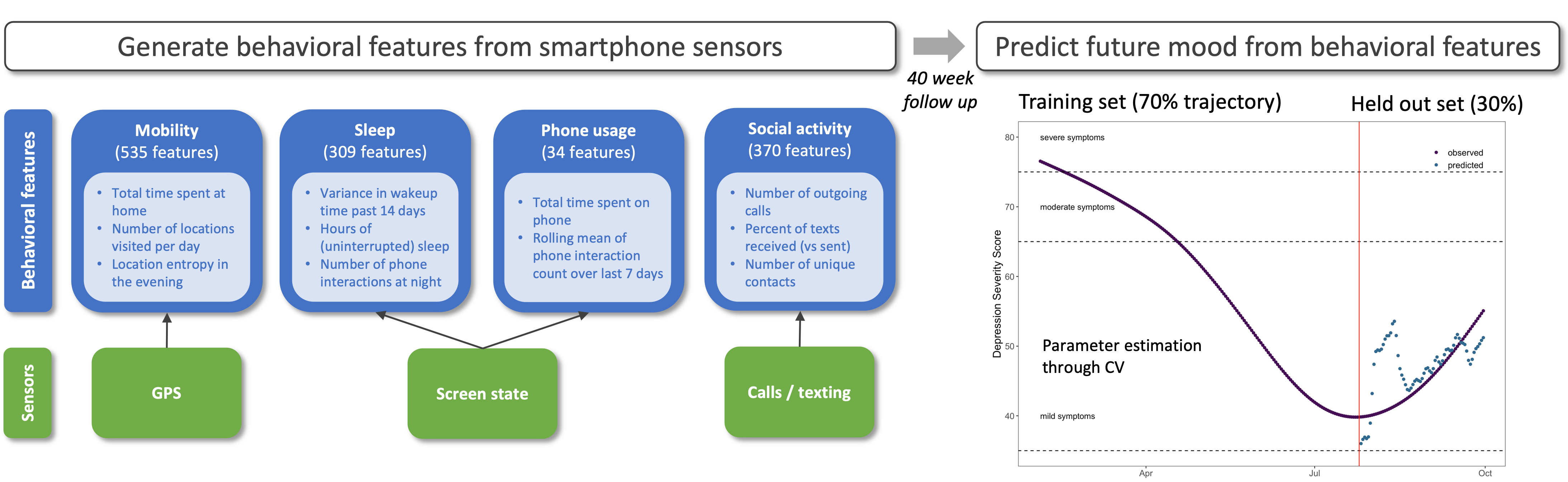
Integrating digital behavioral health tracking in psychiatric genetics research
Genetic studies have now begun to quantitatively assay endophenotypes related to human health and behavior, e.g., sleep and activity levels, by leveraging the vast amount of longitudinal information generated from internet-connected devices, e.g., smartphones and smartwatches. Combining such data with genotype and sequence data will not only enable genetic discovery research (since such endophenotypes might have a strong genetic component) but also help prioritize relevant environmental exposures and behaviors that can module disease risk. This is particularly relevant for psychiatric research, which currently lacks large cohorts phenotyped for quantitative traits. In collaboration with Drs. Nelson Freimer, Jonathan Flint, and Michelle Craske, and as part of the UCLA Depression Grand Challenge, we are developing statistical methods for integrating large, longitudinally collected, digital behavioral health tracking datasets from ethnically diverse cohorts in genetics research of neuropsychiatric disorders.
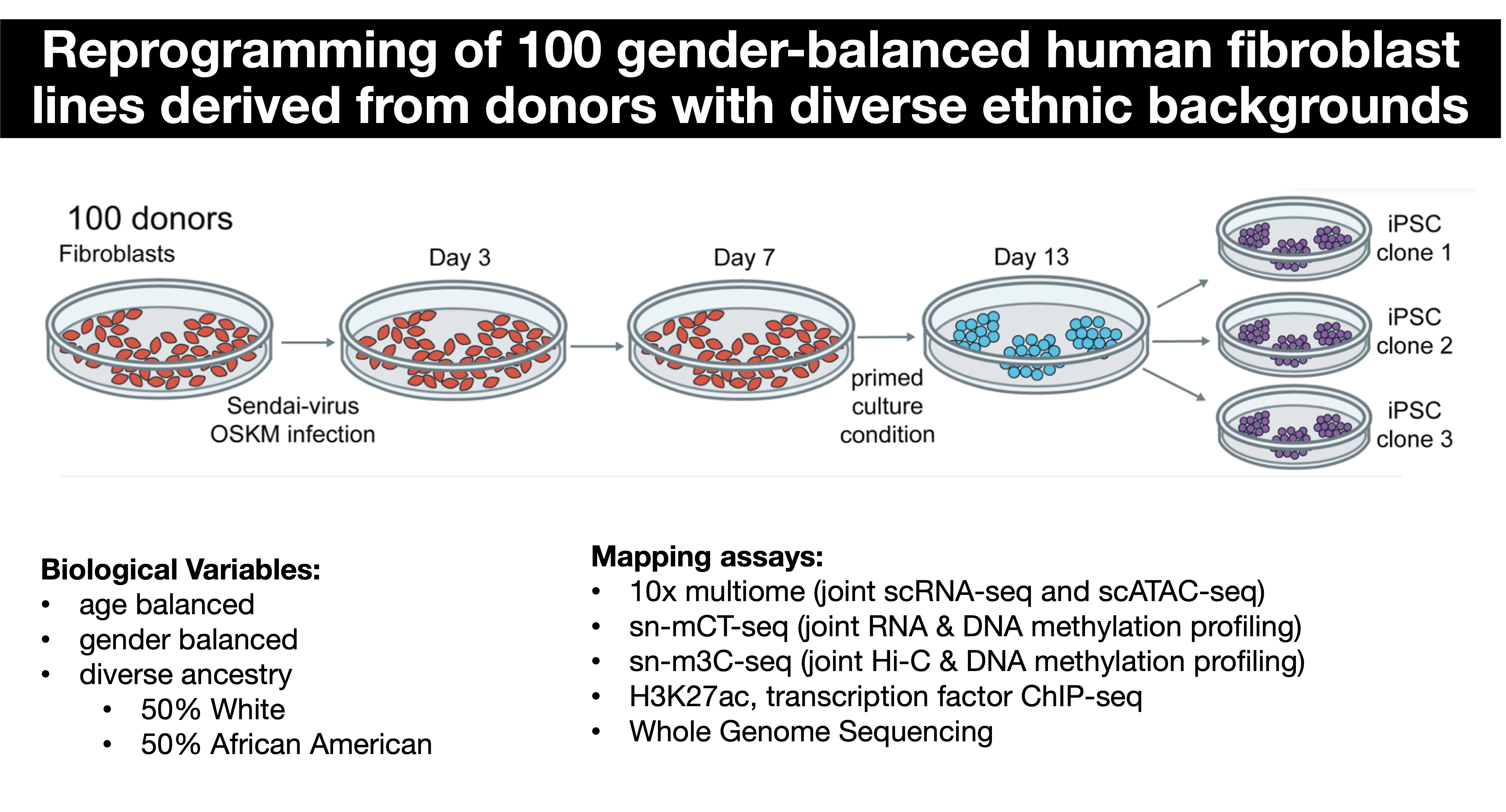
Impact of genomic variation on the gene regulatory networks of reprogramming to pluripotency
The reprogramming of somatic human cells to induced pluripotent stem cells (iPSCs) by only four transcription factors is one of the most striking remodeling of gene regulatory networks. Genetic variation is well known to modulate the regulatory network of pluripotency and contributes to the variability of cellular phenotypes and differentiation capacity of iPSC lines. In collaboration with Drs. Chongyuan Luo and Kathrin Plath, and as part of the IGVF Consortium, we are developing methods to study the impact of diverse population genetics on the gene regulatory networks of reprogramming to pluripotency using population-scale single-cell joint profiling of RNA and DNA methylation and chromatin accessibility. Read more here.
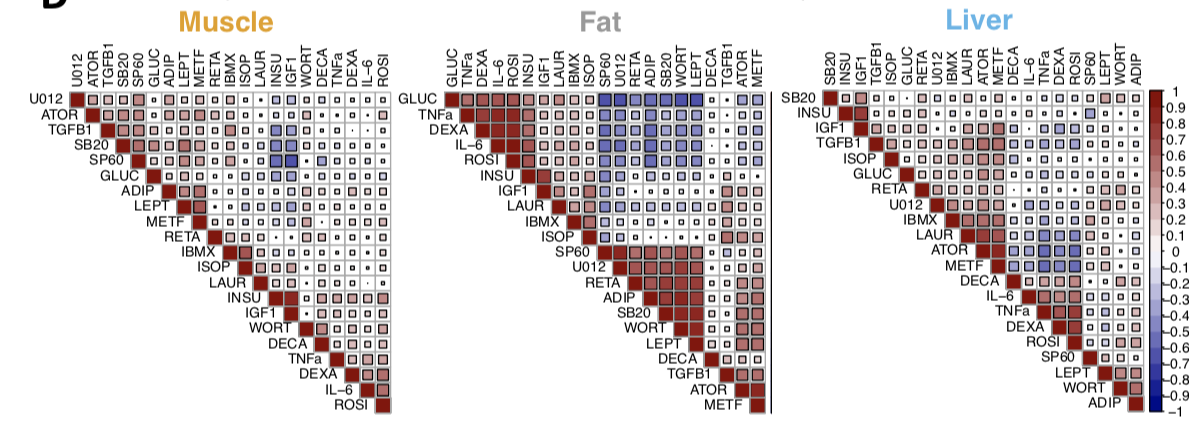
Prioritizing environmental modulators of genetic risk factors for complex traits
Complex traits and diseases can be influenced by both genetics and environment. However, given the large number of environmental stimuli and power challenges for gene-by-environment testing, it remains a critical challenge to identify and prioritize specific disease-relevant environmental exposures. In addition to new statistical methods, we develop combined experimental and computational frameworks for leveraging signals from transcriptional responses to environmental perturbations to identify disease-relevant perturbations that can modulate genetic risk for complex traits (e.g. Balliu et al 2021; AJHG) and inform the functions of genetic variants and genes associated with complex traits (Gloudemans, Balliu et al 2022; Genome Medicine).